Investigator's Brochure Fda Guidance
Investigator's Brochure Fda Guidance - This is an agreement signed by the investigator assuring they will comply with fda regulations related to the conduct of a clinical. 24 this guidance provides recommendations to sponsors and investigators for improving the quality 25 of information they provide to ire3s. Owing to the importance of the ib in maintaining the safety of human subjects in clinical trials, and as part of their guidance on good clinical practice (gcp), the u.s. A comprehensive document that summarizes all available information about a study drug to support clinical research activities, ensuring that. 26 27 fda's guidance documents, including. The fda form 1572 is the statement of investigator. The principles established in this guidance may also be. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data from clinical. Specifically, the guidance provides recommendations for sponsors and investigators conducting investigational new drug (ind) trials to help them differentiate between those adverse events. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. A comprehensive document that summarizes all available information about a study drug to support clinical research activities, ensuring that. Investigator’s brochure.58 a.1 introduction.58 a.2 general. Specifically, the guidance provides recommendations for sponsors and investigators conducting investigational new drug (ind) trials to help them differentiate between those adverse events. 26 27 fda's guidance documents, including. The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. This is an agreement signed by the investigator assuring they will comply with fda regulations related to the conduct of a clinical. You may include a draft version of the protocol. High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. You may include a draft version of the protocol. This section provides guidance to investigators and sponsors (i.e., the responsible parties) on appropriate management of data integrity, traceability and security, thereby. The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. This guidance should be followed when generating clinical trial data that are intended to be submitted to regulatory authorities. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting,. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. The principles established in this guidance may also be. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. The principles established in this guidance may also be. Investigator’s brochure.58. The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. A comprehensive document that summarizes all available information about a. This section provides guidance to investigators and sponsors (i.e., the responsible parties) on appropriate management of data integrity, traceability and security, thereby allowing the. The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. Owing to the importance of the ib in maintaining the. This guidance should be followed when generating clinical trial data that are intended to be submitted to regulatory authorities. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. This section provides guidance to investigators and sponsors (i.e., the responsible parties). The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. The goal of this guidance is to help investigators better meet their. For the most recent version of a guidance, check the fda guidance web page at. The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the. In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme to date, primarily for the benefit of investigators conducting clinical. The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. Investigator’s brochure.58 a.1 introduction.58 a.2 general. Specifically, the. This is an agreement signed by the investigator assuring they will comply with fda regulations related to the conduct of a clinical. The investigator’s brochure (ib) is a compilation of the clinical and nonclinical data on the investigational product(s) that are relevant to the study of the product(s) in human subjects. High quality protocols facilitate proper planning, conduct, reporting, and external review of randomised trials, yet their completeness varies and key elements are often not. Please upload the protocol for the planned study to be submitted to the fda. The food and drug administration issued the final guidance for industry entitled “standardized format for electronic submission of nda and bla content for the planning of. For the most recent version of a guidance, check the fda guidance web page at. In drug development, the investigator’s brochure (ib) summarises the main elements of the entire development programme to date, primarily for the benefit of investigators conducting clinical. Owing to the importance of the ib in maintaining the safety of human subjects in clinical trials, and as part of their guidance on good clinical practice (gcp), the u.s. Investigator’s brochure.58 a.1 introduction.58 a.2 general. The fda form 1572 is the statement of investigator. The principles established in this guidance may also be. You may include a draft version of the protocol. The goal of this guidance is to help investigators better meet their responsibilities with respect to protecting human subjects and ensuring the integrity of the data from clinical. This guidance should be followed when generating clinical trial data that are intended to be submitted to regulatory authorities. Good clinical practice (gcp) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human. The documents reviewed should include the complete documents received from the clinical investigator, such as the protocol, the investigator's brochure, a sample consent.
Investigator Brochure Template Fda

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

PPT What Is An IND? PowerPoint Presentation, free download ID263381

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word

Free Standard Investigator's Brochure Format PDF 3053KB 80 Page(s)

Investigator Brochure Template Fda

Investigator Brochure Template Fda

FREE 10+ Investigator Brochure Templates in AI InDesign MS Word
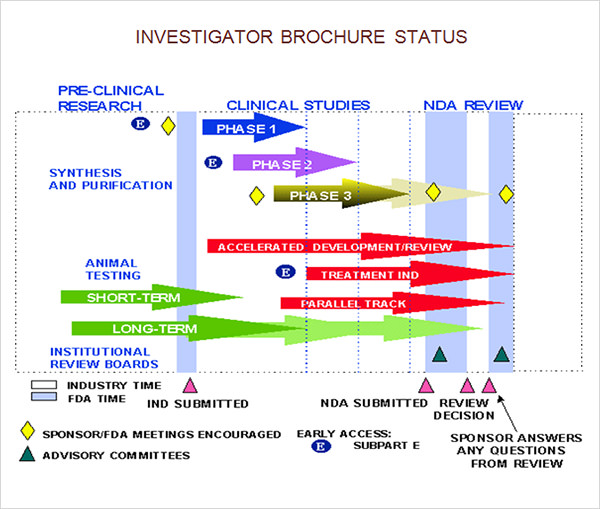
FREE 10+ Investigator Brochure Templates in AI InDesign MS Word
A Comprehensive Document That Summarizes All Available Information About A Study Drug To Support Clinical Research Activities, Ensuring That.
24 This Guidance Provides Recommendations To Sponsors And Investigators For Improving The Quality 25 Of Information They Provide To Ire3S.
This Section Provides Guidance To Investigators And Sponsors (I.e., The Responsible Parties) On Appropriate Management Of Data Integrity, Traceability And Security, Thereby Allowing The.
26 27 Fda's Guidance Documents, Including.
Related Post: